
Today, our manuscript on modular terpene synthesis enabled by mild electrochemical couplings came out in Science! This project was a tremendous journey with lots of ups and downs, successes and setbacks, and many utterings of the phrase “back to the drawing board.” Stephen and I worked side by side on this project for about 3 years. While Stephen was the synthetic mastermind behind the routes and synthetic target selection, I was responsible for developing the electrochemistry needed to pull off the syntheses. There are lots of inventive and creative sequences to a variety of small building blocks in the SI that I implore all interested to check out! His synthetic eye was unlike many people I have seen, and it was amazing to see how he came up with incredibly short routes for some of these building blocks. For this blog post, I wanted to share some of my perspective as the one principally responsible for developing the electrochemistry and how we ultimately arrived at the Ag-nanoparticle solution.
When I joined the project, the tactic that we had originally leveraged to make many of the polyene cyclization precursors was our lab’s decarboxylative alkenylation using redox active esters and vinyl zinc reagents that came out a few years ago. Owing to the reliance on lithium halogen exchange, arduous set up, large excess of reagents, and the need for protecting groups, we elected to branch off into the world of electrochemical reductive coupling to see if that change could alleviate those woes. Reductive coupling couples two electrophiles using a terminal reductant (electricity, or commonly Zn) and a transition metal catalyst. Since no reactive organolithium and organozincs would be generated, more sensitive functionality likely would be tolerated thus shortening synthetic sequences and eliminated the need for protecting groups. Setting up the reaction would also be less cumbersome. No lithium halogen exchanges, transmetallations, titrations, transfers, etc.
After a few months of exploring, we were able to develop some first-generation electrochemical cross coupling conditions that allowed us to couple an expanded range of functional groups including unprotected alcohols. We then got a little ambitious and realized that if we could couple the acids directly on the vinyl iodide piece, then we could conceptually perform coupling after coupling to make linear terpenes and polyenes. However, when we set out to perform a coupling with a vinyl iodide bearing carboxylic acid, the reaction was very low yielding. We tried everything to get this reaction to perform reasonably well with acid bearing vinyl iodides. We explored ligands, electrochemical parameters, solvents, and many other variables all to no avail. We turned to screening electrolytes which is a less desperate way of saying “additive screen.” We tried all sorts of salts, but they only moved the needle a few percent or so. Then we finally found that the addition of AgNO3, initially intended to be an electrolyte, more than doubled the yield of the reaction with the acid bearing vinyl iodides! I ran this experiment just before I left for Thanksgiving and I got this salt from my friend, Samer’s bench where he recently screened electrolytes. In retrospect, I almost didn’t run this experiment had I left for Thanksgiving a day or two earlier and Samer had not just used this salt. This was a truly serendipitous discovery and probably the luckiest I have ever gotten since we were about to seriously reconsider our tactics to pull off the project and go back to the drawing board once again.

Of course, we immediately recognized that silver nitrate is a rather unusual additive for this type of chemistry… after all, silver is usually an oxidant and yet here it is helping in a reductive coupling reaction. It was at this point that we reached out to our collaborators within the CSOE (Center for Synthetic Organic Electrochemistry) to help us understand what role silver could be playing in our reaction.
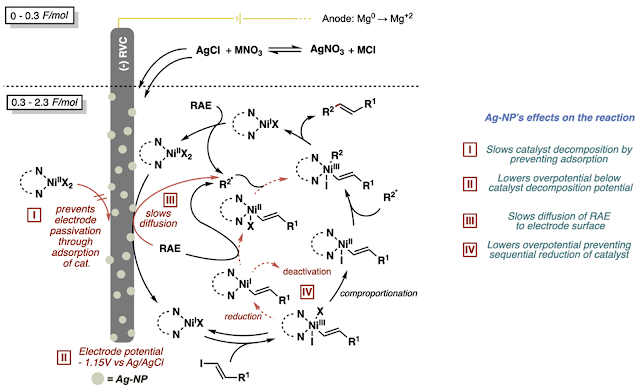
The first observations we made after adding silver to the reaction were 1) the reaction took longer to turn its characteristic red and had an induction period proportional in time to the amount of silver, 2) the electrode was coated in a gray film during the first 0.3 F/mol (read: equivalents of electrons) of the reaction and could be observed after the reaction, 3) the success of the reaction was dependent on having a halide present in that first induction period and 4) the electrode could be reused in a subsequent reaction without needing to add silver nitrate to that second reaction. All these observations led us to think that were first consuming and depositing silver onto the cathode and this newly “functionalized” electrode was responsible for the reactivity boosts we saw.
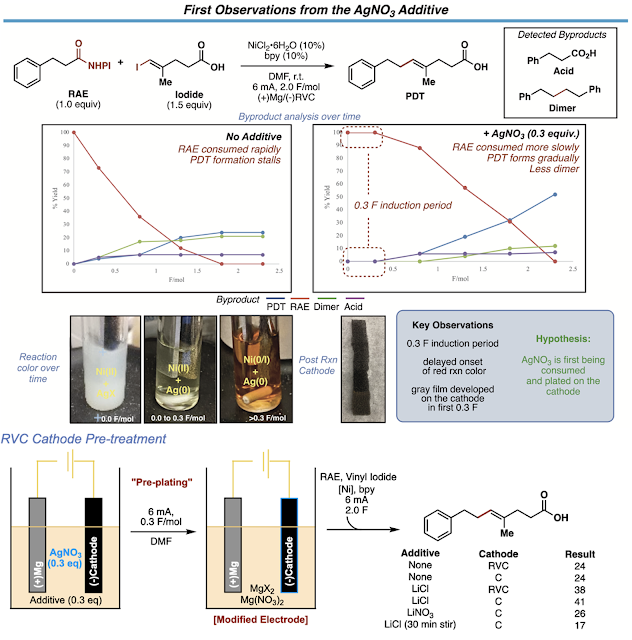
We first teamed up with Dr. Paulo Perez and Professor Scott Anderson at the University of Utah to help us understand what we had done to our electrodes. After sending them many samples of functionalized electrodes (See SI for the workflow and design of these experiments) and sending many emails stating something along the lines of “I think these are the last set of electrodes we want to look at!”, they were able to reveal that in the cases of successful reactions, the electrode surface was littered with silver nanoparticles using S(T)EM imaging and EDX analysis. This was an exciting discovery, but we had absolutely no clue what these particles could be doing in our reaction. After digging around in the literature, it seems such electrodes have been used extensively in analytical sensor design. The deposition of Ag-nanoparticles on carbon electrodes has been used to develop selective sensors for various types of analytes from hydrogen peroxide to nitrobenzene. These sensors operate by either lowering the overpotential necessary for analyte reduction OR by increasing the current response for analyte reduction through nanoparticle-substrate interactions. What had not been reported were cases in electrode decorated with silver nanoparticles support catalytic cross coupling reactions. Go figure.

To start exploring the effect these functionalized electrodes had on the cross-coupling reaction, I first had to find a way to miniaturize the system from our preparative electrodes to an analytic disk electrode that we could use in voltammetry studies. I needed to try to shoot for roughly the same coverage of silver nanoparticles on this smaller surface as we had in the preparative reaction. Doing a little math revealed a good experimental starting point. After a bit of tinkering, I was able to plate silver nanoparticles on the analytical disk electrode reliably just using a dilute solution of Ag and a supporting LiCl additive to ensure we were forming the same type of silver on this electrode as we were in the preparative reaction. Thankfully, anodic stripping experiments, replication of other Ag-NP (silver nanoparticle) based voltammograms, and S(T)EM as well as EDX analysis performed by Paulo helped confirm that we have functionalized our analytic electrode with silver nanoparticles. Time to run some CV’s!
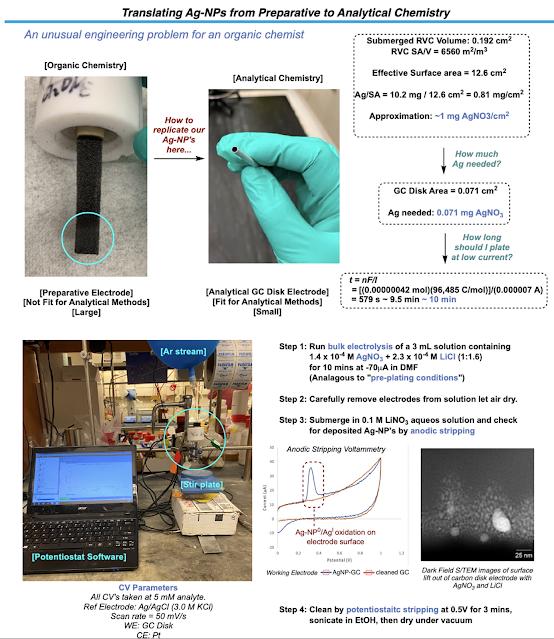
This was probably the most tedious part of my entire PhD. There were tons of components to study in this reaction (catalyst, redox active ester, vinyl iodide, combinations of all of them with and without silver, etc.) which led to me spending weeks at the CV and barely touching synthetic chemistry. To add to this already monotonous undertaking, the analytical electrode (of which I only had one) needed to be freshly plated with the silver nanoparticles each time to ensure the most reliable voltammograms could be produced. We did not want to risk losing any of our silver during analysis. This meant that after every measurement, the electrode had to be oxidatively cleaned until there were no traces of silver, sonicated in EtOH, dried, and then checked again by CV to ensure that all the silver made it off. This long SOP meant that I could only really run 3-4 proper measurements a day and if you go through the SI, I will let you imagine how long it took to get that data. I wish I could say that this led to a watershed in understanding as to what the specific role of silver was but, as chemistry would have it, no dice. The CVs of certain reaction components had very subtle differences with and without silver and nothing that jumped out at us a clear indication of anything spectacular. It was at this point that we sought out our next collaboration.

We teamed up with Professor Héctor Abruña and Cara Gannett at Cornell University to further probe this reaction using some more advanced voltammetry techniques. To make a very long story short, Cara was able to expertly dissect our reaction and its components and study them all using rotating disk electrode (RDE) voltammetry. In very simplified terms, RDE allows you to gain better insight into the kinetics of electrochemical steps of a reaction. I found this website super useful in explaining the technique: https://pineresearch.com/shop/kb/theory/hydrodynamic-electrochemistry/rotating-electrode-theory/.
After a few months, Cara was able to figure out several roles of the silver nanoparticle layer. In summary, the silver basically helps clean up the catalytic cycle for the cross-coupling reaction. It prevents catalyst degradation/deactivation (which is especially prevalent when using vinyl iodides bearing unprotected acids) and prevents background consumption of redox labile functionality such as the redox active ester. You can check out the paper and the SI for more!
After having made this truly serendipitous discovery and working for nearly 2 years to understand the finding, the project was brought back from the brink and was making a turn towards what we set out to accomplish: a robust platform for terpene synthesis. After demonstrating that the silver additive now allowed the reaction to tolerate free acids as well as many other functional groups, we shifted our attention to developing an in-situ protocol so that we could skip a step in redox active ester preparation. We quickly found some very simple conditions (acid, NHPI, DIC, THF, 1hr) and we were off to the races. Together, we made 13 molecules that highlight the tolerance of various functional groups and demonstrate the plug and play nature of this synthetic strategy. I won’t talk too much about the synthesis as they are discussed in detail in the paper and SI but I would like to shout out Asymchem for demonstrating that both the generation of silver nanoparticles and the electrochemical coupling could be performed on 100g scale in recirculating flow! Thank you Zhen and Lijie for your time and expertise!
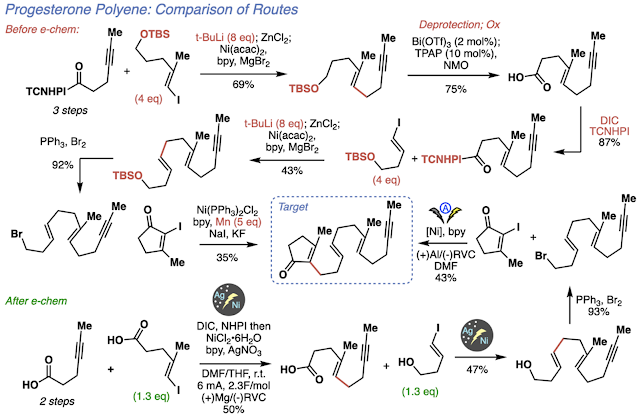
Overall, the tactics of this project came a long way from using alkylithiums and long arduous set ups. Now, with the silver-nanoparticle functionalized electrodes and the power of electrochemistry, we were able to run this chemistry in a true dump and stir fashion to make polyenes in a streamlined fashion. Just to highlight how much electrochemistry changed the project, the previous route developed towards the progesterone polyene required a large amount of pyrophoric reagents for the cross couplings, tedious procedures, protecting groups and intermediate functional group interconversions. Using electrochemistry and after developing the necessary tactics, we could make the same polyene in fewer steps, with no protecting groups, and in an operationally simple fashion. We recognize that this reaction sounds deceptively super hard to run. It has nickel, electrochemistry, and nanoparticles, it sounds like a nightmare. However, I assure you that it is so easy that even Phil and his “dilapidated hands” can do it:
If I haven’t made it totally clear yet, this project was a true team effort. No single person could have pulled this off single handedly. Paulo and Scott were instrumental in our electrode surface understanding and their expertise in microscopy was invaluable as they revealed the crucial formation of silver nanoparticles. Héctor and Cara brought us to understand the mechanistic underpinnings of the reaction and demystified the role of the nanoparticle functionalized electrode. Zhen and Lijie showcased the scalability of the platform in an awe-inspiring manner. Phil granted us grace, guidance, and patience as we have been telling him that were almost done for nearly 2 years… Finally, Stephen initiated this project a year before I joined and has been a great mentor to me. He taught me a lot about synthesis, and I am very grateful for his patience and generosity in letting a first year come into the project and try to stick electrodes in it.
So anyway, that’s the story about how a lucky hit in an additive screen resulted in the story we are blessed to be able to tell today.
-Max, on behalf of Team Polyene.





























